
今天推送的文章为发表在Angew.Chem.Int.Ed.的“Asymmetric C1Extension of Aldehydes through Biocatalytic Cascades for Stereodivergent Synthesis of Mandelic Acids”。
手性α-羟基羧酸是各种生物活性分子中普遍存在的基序之一,也是不对称合成中有价值的组成部分。对映体纯扁桃酸盐及其衍生物因其在有机和药物化学中的多用途应用而备受关注(图 1a)。目前已开发了许多合成外消旋扁桃酸酯的分步工艺,而扁桃酸酯的不对称合成依赖于相应酮酸的金属依赖性氢化。然而,所有这些方法都需要昂贵有毒的试剂、苛刻的反应条件,造成浪费。
生物催化级联为扁桃酸酯的生产提供了更环保、更可持续的程序。扁桃酸盐的常规生物催化生产是通过脂肪酶催化的动力学拆分或严格立体选择性的羟基腈裂解酶和腈水解酶的组合来完成(图 1b)。然而,这些酶促方法需要剧毒氰化物来进行生物转化本身或制备底物。有研究报导了一种以硫胺素二磷酸依赖性碳连接酶为中心的策略,用于体外从苯甲醛和草酸盐生产(S)-扁桃酸酯(图 1c),这是迄今为止报道的唯一一条无氰化物C1扩建路线,但需要外部ATP和辅酶A。
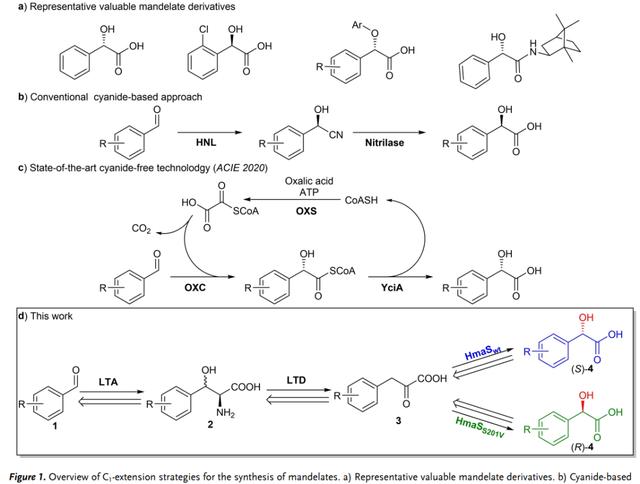

在本研究中,作者报道了从苯甲醛立体发散合成(S)-和(R)-扁桃酸酯衍生物的新的生物催化级联反应(图 1d)。该级联反应不需要外部氧化还原辅因子或有毒试剂氰化物,只需要苯甲醛、大量的甘氨酸和空气中的O2作为化学试剂,显示出良好的原子经济性。该级联反应具有广泛的底物范围、效率高(产量高达98 %), 具有优异的对映选择性(最高>99 % ee),并能够应用于克级生产。
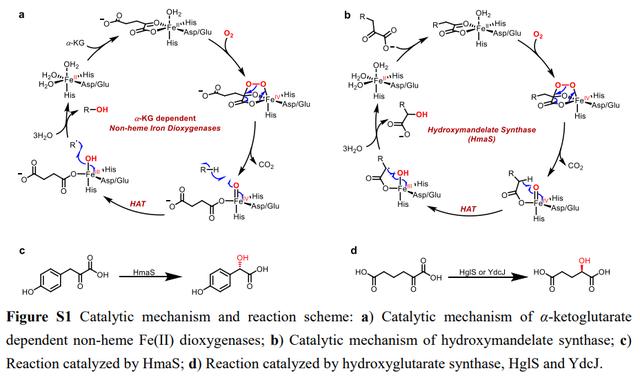
作者最初受到一种特殊类型的酮酸依赖性非血红素FeII双加氧酶独特催化机制的启发。与α-酮戊二酸依赖性双加氧酶的经典催化机制不同(图 S1a),这些加氧酶催化α-酮酸与分子氧氧化脱羧后Cβ-H位的羟基化,而不是第二底物(图 S1b)。例如,(S)-4-羟基扁桃酸合成酶(Hmass)将4-羟基苯基丙酮酸转化为(S)-4羟基扁桃酸酯,这是(S)-4-羟基苯基甘氨酸生物合成的关键步骤(图 S1c)。羟基戊二酸合成酶(HglS和YdcJ)负责植物和细菌赖氨酸分解代谢的最后一步,从2-氧代己二酸中产生(R)-羟基戊二酸(图 S1d)。
利用逆向生物合成的逻辑,作者设想这些加氧酶可以与低特异性的L-苏氨酸醛缩酶(LTA,EC 4.1.2.48)和L-苏氨酸脱水酶(LTD,EC 4.3.1.19)合作,从苯甲醛产生扁桃酸酯,导致立体发散的不对称C1延伸转化。该生物合成途径的第一个反应是甘氨酸和苯甲醛在LTA存在下的羟醛反应(图 1d)。LTA产生的中间体苯基丝氨酸可以通过LTD转化为相应的非手性α-酮酸,然后通过加氧酶脱羧和立体发散不对称C-H键羟基化,得到所需的扁桃酸酯。LTD和双加氧酶催化的反应在热力学上非常有利,并作为级联反应形成产物的驱动力。
为了实现所提出的级联反应,作者首先鉴定了用于脱羧羟基化的有效的加氧酶。重组表达和纯化AoHmaSwt和一个突变体(AoHmaSS201V)用于生产(S)-扁桃酸盐,使用来自P.putida的HglS和来自E.coli的YdcJ的突变体生产(R)-扁桃酸盐。在标准条件下用苯基丙酮酸盐测定所有纯化的酶。AoHmaSwt和AoHmaSS201V表现出高活性,产生超过98%的扁桃酸盐,而HglS和YdcJ的活性较低,产生的扁桃酸少于10%。手性HPLC分析表明,AoHmaSwt产生了具有天然对映选择性的(S)-扁桃酸盐,ee为73%,而AoHmaSS201V表现出相反的对映选择性,以99% ee产生(R)-扁桃酸盐。
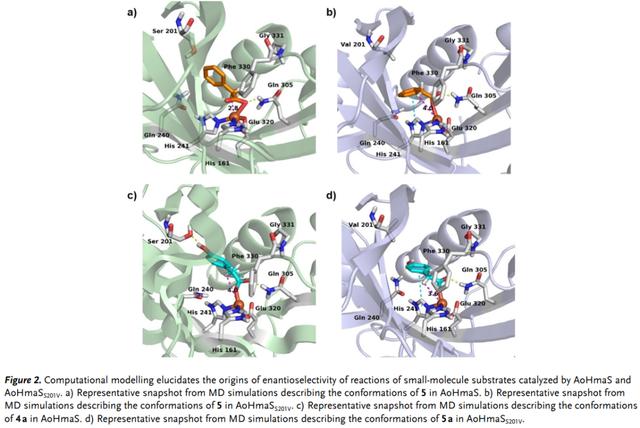
为了深入了解AoHmass和AoHmaSS201V的对映选择性反转,作者分别对其与中间体苯乙酸(5)和对羟基苯基乙酸(5a)的复合物进行了分子动力学(MD)模拟。沿MD轨迹测量Fe-H距离,以估计与底物的对映选择性。在AoHmaS中,无论是5还是5a,Fe和pro-(S)-H在Cα-位置的距离比pro-(R)-H更接近。代表性的稳定构象如图所示 2a和c,其中Fe中心和Cα位置的pro-(S)-H之间的距离分别为2.8和4.0 ?。AoHmaSS201V的情况有所不同。5和5a的芳香环发生翻转并在 MD模拟25 ns后保持稳定。翻转的5和5a在AoHmaSS201V中与His241的π–π相互作用增加。这种相互作用和Val201的空间位阻可能是5和5a构象变化的驱动力。与AoHmaSwt中的5和5a相比,AoHmaSS201V中的5和5a的构象翻转导致Fe和pro-(R)-H之间的距离更近,分别为4.0和3.6 ?(图 2b,d),有利于(R)-扁桃酸的形成。
在筛选得到S-和R-选择性扁桃酸合成酶的情况下,作者开始鉴定用于羟醛反应和脱水的LTA和LTD。在第一步中,选择来自P.putida的苯基丝氨酸醛缩酶(PpLTA)作为苯甲醛和甘氨酸之间的醛缩反应的酶。在级联反应的第二步中,苏氨酸脱水酶RpLTD、PxLTD或CgLTD被用作苯基丝氨酸的脱水和水解的酶,这些LTD已被证明能催化所提出的反应。然后进行了体外级联反应,以研究这些选定酶的活性和兼容性。纯化的PpLTA与RpLTD和PxLTD组合在体外显示出很高的效率,并将苯甲醛转化为苯丙酮酸,转化率>95%。随后,包括PpLTA、RpLTD和AoHmaSwt在内的体外级联反应以98%的速率在2 h成功地从苯甲醛中生产了扁桃酸。
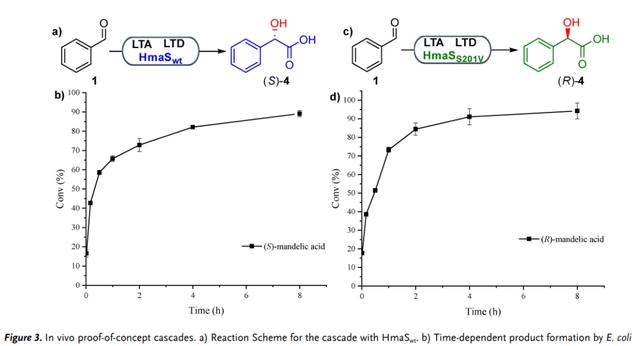
然后,作者试图将体外级联组装到大肠杆菌中进行全细胞生物转化。结果发现 15mg mL?1携带pACYC-Duet_pLTA_RpLTD和pET-22b_AoHmaSw的冻干大肠杆菌细胞,在8 h时以89% 的转化率生成(S)-扁桃酸(图3a、b)。携带pACYC-Duet_pLTA_RpLTD和pET-22b_AoHmaSS201V的大肠杆菌细胞在8 h时以94% 的转化率生成(R)-扁桃酸(图3c、d)。
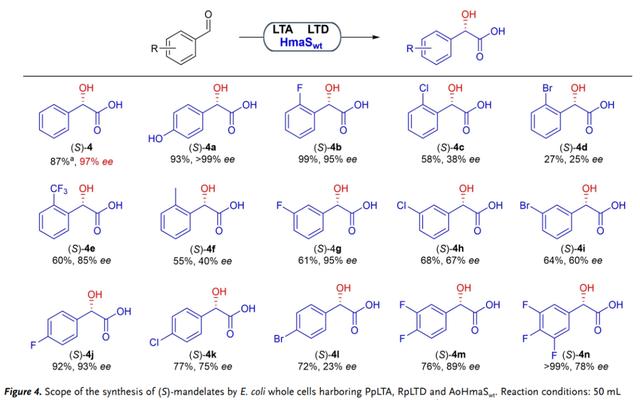
接下来,作者试图证明级联在扁桃酸盐衍生物生产中的稳健性和合成效用。首先,使用AoHmaSwt的全细胞催化剂将具有取代基的苯甲醛转化为预期产物(图 4, 4a–4n),产率为39–93%和ee值为25-99%。在所有产品中,对羟基扁桃酸盐(4a),HmaS的天然底物,以93%的最高产量和>99%的ee生产;间氟和对氟扁桃酸酯(4g和4j)以高产率和对映选择性生产。对于具有其他取代基的底物,效率和选择性显著降低(产物4c–4f、 4h–4n)。
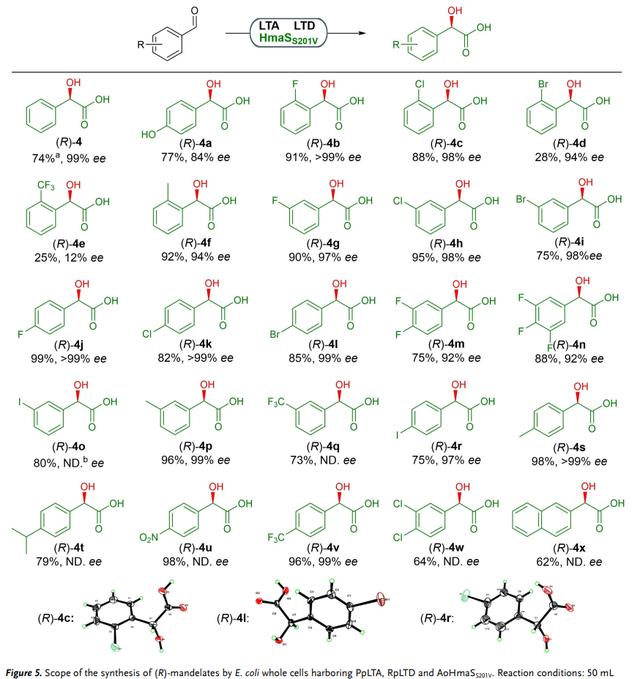
相比之下,具有AoHmaSS201V的全细胞催化剂显示出更广泛的范围,能接受在所有位置具有不同取代基的底物(图 5,4–4x)。该催化剂对在不同位置具有甲基、氟、氯和溴取代基的苯甲醛以优异的产率和对映选择性转化(4–4c、 4f–4l、 4o–4v),对o位有较大的吸电子取代基底物的催化效率较低,导致降产率(4d)或低对映选择性(4e)。此外,对二-和三氟取代的底物也以高产率和对映选择性转化(4m和4n)。
最后,作者进行了600mL的克级级联反应以生产抗血小板药物氯吡格雷的关键中间体(R)-2-氯-苯二酸(4c), 产率为81%(0.91 g)。
创业项目群,学习操作 18个小项目,添加 微信:luao319 备注:小项目!
如若转载,请注明出处:https://www.fqkj168.cn/7773.html
相关推荐
-
抖音短视频剪辑怎么赚钱,抖音短视频剪辑怎么赚钱_在抖音搞钱的小妙招分享
抖音电影,昨天收益:581.82。新手入门教程。 昨天晚上在抖音看了一场电影,赚了有500多块。你知不知道最近抖音新出了一个赚钱的玩法,反正在家里呆着也没事,我就只能去追剧。没想到…
-
北大纵横,北大纵横管理咨询集团
北大纵横管理咨询集团是中国管理咨询领域的领军者之一,成立于1994年,总部位于北京。该集团是由北京大学光华管理学院的教授和学者们共同创立的,旨在将学术研究成果与实践经验相结合,为企…
-
孩子视力4.7是近视多少度,8岁孩子视力4.7是近视多少度
近视度数是指眼球长短和水晶体屈光度之间的比例关系。视力4.7是一种常见的近视度数,会导致孩子眼睛对远处的物体看不清楚。那么,对于8岁孩子来说,视力4.7到底是近视多少度呢?接下来,…
-
两男一女爱爱,两男一女p3
近?年来,社?风会?气和?们人?的精?状神?态?发愈?复杂?多变,人们?生的?活方式、价值?念观?也在?然悄?转变。面对众?不多?良风气,家?们长?挂心?孩的?子们也?现出?了越?…
-
青少年近视防控中心是干嘛的,青少年近视防控中心是干嘛的呢
青少年近视防控中心是干嘛的,青少年近视防控中心是干嘛的呢?这个问题不仅关乎着家长们对孩子视力健康的担忧,也牵动着全社会对青少年健康成长的关切。近年来,青少年近视问题日益凸显,为了有…
-
济宁哪家做包皮手术医院最好,济宁哪家做包皮手术医院最好的
济宁瑞康医院:创新发展让医疗服务便捷高效 近年来,随着医疗服务的不断提升和医疗技术的快速发展,越来越多的民营医院开始崛起。济宁瑞康医院作为山东省济宁市的一家民营医院,不仅为当地居民…
-
地锅鸡的制作方法,地锅鸡的制作方法视频教程
地锅鸡,作为一道美味的家常菜,深受人们喜爱。它的口感鲜嫩多汁,香味浓郁,回味无穷。想要在家享用到正宗的地锅鸡,掌握正确的制作方法至关重要。在这篇文章中,我们将一起学习制作地锅鸡的方…
-
付费软件,付费软件怎么退订
随着科技的不断发展,我们的生活越来越离不开各种软件。而有些软件可能需要付费才能使用,但是在使用过程中,我们可能会发现这些软件并不符合我们的需求,或者我们不再需要它们了。这时候,我们…
-
rser期刊,rser期刊怎么样
植物和食果动物相互作用在维持森林生态系统的生物多样性上扮演着重要的角色。由于果实资源在空间上呈垂直分布,同时动物在垂直空间利用上也存在分化,食果网络在森林群落尺度上也会呈现垂直结构…
-
摇摇鞋真的能瘦腿吗,摇摇鞋真的能瘦腿吗知乎
实测网络热门产品:网红摇摇鞋。 商家说穿上这个鞋既能瘦身还能美腿,来先看卖家秀。你腿这么好看怎么练的?就是穿的这款摇摇鞋。我试一下,感觉怎么样?这个腿好酸。当然了每天坚持5分钟,可…